
OCR Specification focus:
‘Outline the principles of DNA sequencing and summarise advances from Sanger methods to modern high-throughput techniques.’
DNA sequencing reveals the precise order of nucleotides—adenine (A), thymine (T), cytosine (C), and guanine (G)—within a DNA molecule, providing crucial insight into gene function, variation, and evolution. This process has evolved dramatically, from the early Sanger sequencing methods of the 1970s to today’s high-throughput next-generation sequencing (NGS) platforms that can sequence entire genomes rapidly and affordably.
Principles of DNA Sequencing
The Concept of DNA Sequencing
DNA sequencing determines the exact nucleotide sequence within a DNA fragment. It allows scientists to decode genetic instructions, compare genomes, and identify mutations responsible for genetic diseases.
DNA Sequencing: The laboratory technique used to determine the exact order of nucleotides (A, T, C, G) in a strand of DNA.
Sequencing relies on DNA polymerase, an enzyme that synthesises complementary DNA strands by adding nucleotides in the 5’ to 3’ direction. By analysing which nucleotide is incorporated at each step, the sequence of the original DNA can be deduced.
Key Components in DNA Sequencing
Template DNA: The DNA strand to be sequenced.
Primer: A short strand of nucleotides that initiates DNA synthesis.
DNA Polymerase: Enzyme responsible for adding nucleotides to the growing strand.
Deoxynucleotides (dNTPs): Normal nucleotides (A, T, G, C) used to synthesise DNA.
Dideoxynucleotides (ddNTPs): Modified nucleotides lacking a 3’-OH group, which terminate chain elongation when incorporated.
The Chain-Termination Principle
The chain-termination principle, first developed by Frederick Sanger in 1977, is based on the incorporation of dideoxynucleotides during replication.
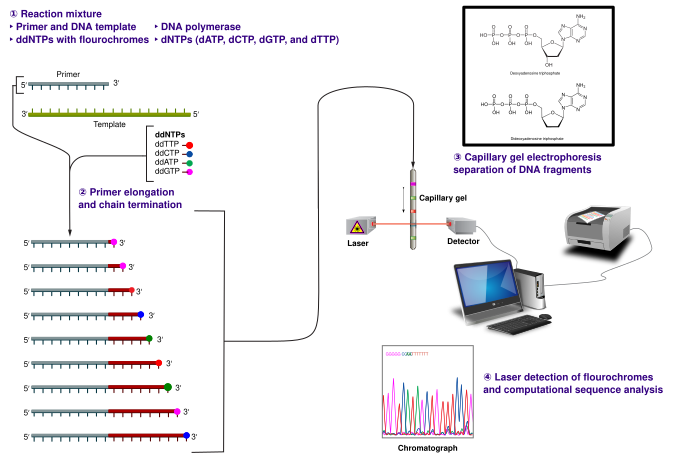
Sanger sequencing uses a primer, DNA polymerase, normal dNTPs, and rare ddNTPs to terminate strand extension at specific bases. Terminated fragments of all lengths are separated by capillary electrophoresis and read by fluorescence. The diagram shows each major stage in order. Source.
Dideoxynucleotide (ddNTP): A nucleotide analogue lacking the 3’-hydroxyl group necessary for phosphodiester bond formation, causing termination of DNA synthesis.
When these fragments are separated by size, the original sequence can be reconstructed by reading the final nucleotide at the end of each fragment.
The Sanger Sequencing Method
Traditional Sanger Sequencing Process
The Sanger (chain-termination) method involves several key stages:
DNA denaturation: The double-stranded DNA is separated into single strands.
Primer annealing: A primer binds to the template strand.
DNA synthesis: DNA polymerase extends the strand using dNTPs and occasional ddNTPs labelled with a fluorescent tag.
Chain termination: Random incorporation of ddNTPs halts synthesis at various points.
Separation by electrophoresis: The resulting fragments are separated by capillary electrophoresis, where smaller fragments migrate faster through a gel or capillary tube.
Detection: A laser and detector read the fluorescently labelled ddNTPs to determine the sequence order.
Output and Accuracy
The output from Sanger sequencing is a chromatogram, showing peaks corresponding to the fluorescent signals of each nucleotide.
While highly accurate, this process is slow and labour-intensive, typically reading only around 700–1000 base pairs per reaction.
Advances to High-Throughput Sequencing
Need for Faster Techniques
As genome projects expanded, traditional Sanger sequencing became impractical for sequencing large genomes such as the human genome, which contains approximately 3 billion base pairs. This led to the development of high-throughput sequencing technologies, collectively known as Next-Generation Sequencing (NGS).
Principles of Next-Generation Sequencing (NGS)
NGS technologies apply the same core principle—identifying nucleotides as they are incorporated—but automate and parallelise the process. Millions of fragments are sequenced simultaneously, dramatically increasing output.
Main Stages of NGS:
DNA fragmentation: The genome is broken into millions of small fragments.
Adaptor ligation: Short DNA sequences (adaptors) are attached to fragment ends to facilitate binding to a sequencing platform.
Amplification: Each fragment is clonally amplified, often on a solid surface such as a flow cell or within tiny beads.
Sequencing by synthesis: Fluorescently labelled nucleotides are incorporated one at a time, and a camera captures the emitted fluorescence to record which base has been added.

Clusters on a flow cell are imaged cycle-by-cycle as reversible terminator nucleotides are incorporated, with colour revealing the next base. Millions of clusters are read in parallel to achieve high throughput. The minimalist schematic focuses on the core principle; platform-specific chemistry details are beyond the OCR syllabus. Source.
Data processing: A computer compiles the data to reconstruct the full DNA sequence.
Types of High-Throughput Sequencing Technologies
Illumina (sequencing by synthesis): Uses reversible terminator nucleotides and imaging of incorporated bases. It offers extremely high accuracy and throughput.
454 Pyrosequencing (now discontinued): Detected light emitted by pyrophosphate release when a nucleotide was incorporated.
Ion Torrent: Detects hydrogen ions released during nucleotide addition, converting chemical changes into electrical signals.
Oxford Nanopore and PacBio (third-generation): Sequence single molecules of DNA directly, producing long reads that aid in mapping repetitive or complex regions.
Advantages of Modern Sequencing
Massive parallel processing: Millions of sequences generated in a single run.
Lower cost per base: Whole-genome sequencing is now affordable for research and clinical use.
High speed: A human genome can be sequenced within days instead of years.
Comprehensive coverage: Detects mutations, insertions, deletions, and variations across entire genomes.
Challenges and Limitations
Despite advances, NGS also presents challenges:
Data management: Enormous data volumes require powerful computational tools for storage and analysis.
Error rates: Some methods have higher raw error rates than Sanger sequencing.
Short read lengths: Many platforms produce short reads that complicate assembly, though third-generation methods are improving this.
Ethical concerns: The ease of sequencing raises privacy issues regarding genetic information.
The Transition from Sanger to Modern Techniques
The shift from Sanger sequencing to NGS represents a major technological leap:
Sanger sequencing: Ideal for small-scale, highly accurate sequencing tasks such as confirming mutations.
NGS: Designed for large-scale genome analysis, transcriptomics, and metagenomics.
Together, these methods underpin modern genomics, molecular diagnostics, and bioinformatics, allowing detailed exploration of life at the molecular level and fulfilling the OCR specification aim to understand both the principles of sequencing and the advances that enable high-throughput analysis.
Practice Questions
Question 1 (2 marks)
Describe how the incorporation of dideoxynucleotides (ddNTPs) leads to chain termination during Sanger DNA sequencing.
Mark Scheme:
1 mark: States that ddNTPs lack a 3’-hydroxyl (OH) group on the deoxyribose sugar.
1 mark: Explains that without a 3’-OH group, no further nucleotides can form phosphodiester bonds, stopping DNA strand elongation.
Question 2 (5 marks)
Explain how modern high-throughput sequencing techniques differ from the traditional Sanger method, and discuss two advantages of these modern approaches.
Mark Scheme:
1 mark: States that high-throughput sequencing (next-generation sequencing) sequences millions of fragments simultaneously, whereas Sanger sequences one fragment at a time.
1 mark: Describes that NGS uses automated, parallel sequencing by synthesis with fluorescently labelled nucleotides and imaging.
1 mark: States that Sanger sequencing is slower and more labour-intensive.
1 mark: Mentions an advantage such as increased speed, allowing whole genomes to be sequenced within days.
1 mark: Mentions another advantage such as lower cost per base, greater data output, or broader genome coverage.
FAQ
The key difference lies in the sugar structure. ddNTPs lack the 3’-hydroxyl (–OH) group on the deoxyribose sugar that normal dNTPs possess.
Without this group, a phosphodiester bond cannot form with the next nucleotide, halting chain elongation.
This chemical difference is what allows ddNTPs to terminate DNA synthesis at specific points, a fundamental principle of Sanger sequencing.
Earlier sequencing methods used radioactive isotopes to identify nucleotides, but fluorescent labels are now preferred for safety and efficiency.
Fluorescent dyes emit light at specific wavelengths corresponding to each base (A, T, C, G).
This allows real-time detection through laser excitation and imaging.
Fluorescence avoids radioactive waste and enables automated sequencing by computer-controlled systems.
Even if multiple fragments terminate at the same base, they differ in length, and separation by capillary electrophoresis resolves these differences.
Each fragment migrates according to its size — shorter fragments move faster through the gel matrix.
The detector identifies the fluorescent tag on the terminating base of each fragment, ensuring accurate sequence reconstruction from smallest to largest.
Bioinformatics processes and interprets the vast data produced by high-throughput sequencing.
Key functions include:
Sequence assembly: Aligning millions of short reads into continuous genomic sequences.
Error correction: Identifying and correcting sequencing inaccuracies.
Annotation: Locating genes and regulatory regions within the genome.
Data storage: Managing terabytes of raw sequence data efficiently.
The Human Genome Project (completed in 2003) cost around $3 billion and took over a decade.
Modern high-throughput sequencing can now sequence a human genome for under £1,000 in a few days.
This dramatic cost reduction results from automation, miniaturisation, and massively parallel processing, making genome sequencing accessible for medical diagnostics, evolutionary studies, and personalised medicine.
