
OCR Specification focus:
‘Predict carbon-13 or proton NMR spectra for given molecules studied in the course.’
Introduction
Predicting carbon-13 and proton NMR spectra is essential for understanding how molecular structure determines the number, type, and behaviour of chemical environments. These notes explain how to anticipate key NMR features using structural reasoning.
Predicting NMR Spectra
Accurate prediction of NMR spectra requires understanding how different atoms and functional groups influence chemical shift, integration, and splitting patterns. OCR expects students to predict both ¹³C NMR and ¹H NMR spectra for molecules studied in the course by analysing the number of environments and the expected chemical-shift regions.
Key Principles in NMR Prediction
When predicting spectra, the central questions are:
How many unique chemical environments are present?
What chemical-shift region will each environment appear in?
What will the relative intensity (integration) be?
For ¹H NMR, how will neighbouring protons influence spin–spin splitting?
Before applying these principles, students must understand the term chemical environment.
Chemical environment: The distinct electronic surroundings of a nucleus, leading to a unique NMR signal when not chemically or symmetrically identical to others.
Normal sentences separate definition blocks to maintain clarity and flow.
Predicting ¹³C NMR Spectra
¹³C NMR spectra show a separate peak for each carbon environment, without splitting under standard proton-decoupled conditions. This makes carbon spectra relatively straightforward to predict.
Determining the Number of Carbon Environments
The first step is identifying how many carbons are chemically distinct.
To do this, consider:
The presence of symmetry elements in the molecule
Whether carbons are attached to different atoms or groups
Changes in hybridisation (e.g., sp³, sp², sp)
Anticipating ¹³C Chemical Shift Regions
Carbon environments can be associated with broad chemical-shift regions:
0–50 ppm: saturated sp³ carbons not bonded to electronegative atoms
50–100 ppm: sp³ carbons bonded to heteroatoms, alkynes
100–160 ppm: alkenes and aromatic carbons
160–220 ppm: carbonyl carbons (C=O)
Chemical shifts vary with electronegativity and conjugation. For example, carbons attached to oxygen shift further downfield.
Predicting ¹H NMR Spectra
¹H NMR spectra provide richer detail because protons exhibit chemical shift, integration, and splitting through interaction with neighbouring protons.
Identifying Proton Environments
As with carbons, the number of proton environments depends on molecular symmetry and connectivity. Equivalent protons produce one signal.
Predicting ¹H Chemical Shifts
The predicted shifts depend on functional groups:
0.5–2.0 ppm: alkyl protons
2.0–3.0 ppm: protons adjacent to electronegative atoms
4.0–6.0 ppm: protons on double bonds (alkenes)
6.0–9.0 ppm: aromatic protons
9.0–10.0 ppm: aldehydic protons
These ranges should be used as guidance when predicting spectra. Highly electronegative substituents shift signals downfield because they withdraw electron density.
Integration and Proton Ratios
¹H integration indicates the relative number of protons contributing to each signal.
It is essential for determining structural fragments and must correlate with proton counts predicted from the molecular formula.
Integration (NMR): A measure of the relative area under an NMR signal corresponding to the number of equivalent protons contributing to that environment.
A normal sentence ensures the spacing rule between definitions.
Predicting Splitting Patterns
The n + 1 rule states that a proton with n equivalent neighbouring protons splits into n + 1 peaks. Splitting only occurs between protons on adjacent carbon atoms unless restricted by structural factors.
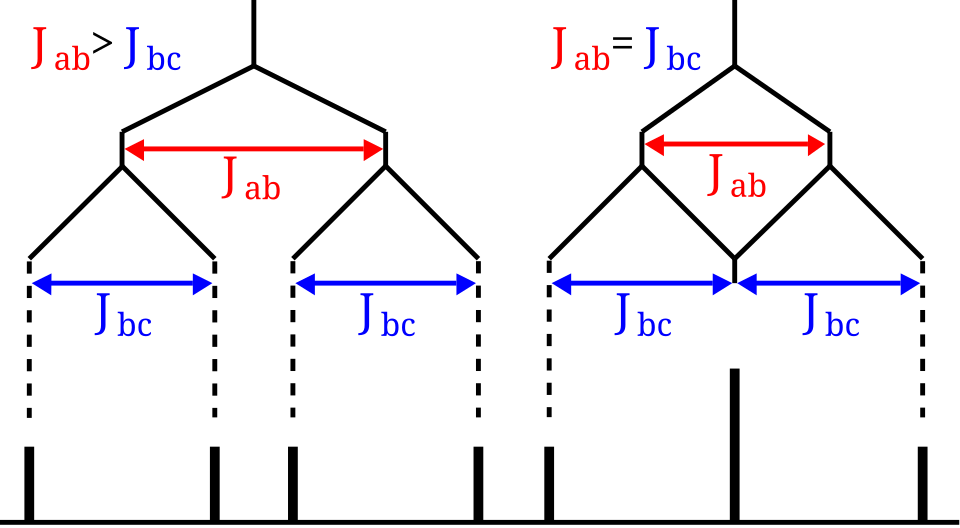
This splitting tree shows how spin–spin coupling produces characteristic multiplets and intensity ratios such as 1:2:1. The left-hand example includes a doublet of doublets, which extends beyond simple n + 1 cases but remains useful for prediction. Source
Key points:
Singlet: no neighbouring protons
Doublet: 1 neighbour
Triplet: 2 neighbours
Quartet: 3 neighbours
Larger multiplets indicate several non-equivalent neighbours
Coupling does not occur when protons exchange rapidly (e.g., O–H or N–H in the presence of acid or moisture).
Combining Predictive Elements
When producing a predicted spectrum for a molecule studied in the course, combine all three elements:
Number of environments (unique carbons or protons)
Expected shift range (functional group influence)
Intensity or splitting (¹H only)
Each peak in a predicted spectrum represents one distinct ¹H or ¹³C environment, so the first step is always to count chemically different nuclei (using symmetry where possible).
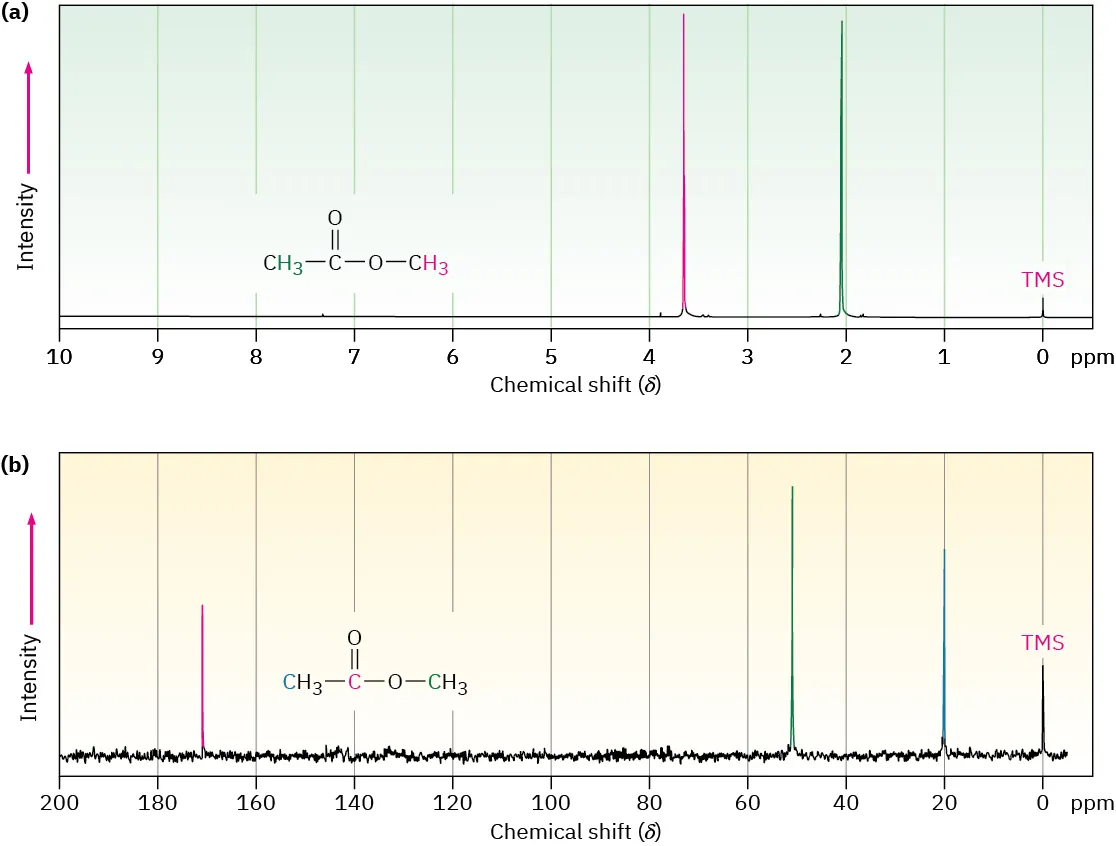
This paired spectrum compares ¹H and proton-decoupled ¹³C NMR for the same molecule, illustrating how proton equivalence reduces the number of signals while each carbon environment gives a separate peak. Source
Structural Features that Influence NMR Predictions
Consider the following influences during prediction:
Electronegative atoms
Withdraw electron density, pushing signals downfield
Broaden shift ranges for both ¹³C and ¹H
Hybridisation
sp² carbons resonate further downfield than sp³
Aromaticity
Ring currents generate characteristic downfield shifts
Conjugation
Delocalisation alters electron density and shifts
Symmetry
Limits the number of distinct environments
Functional groups
Carbonyls, alcohols, amines, and aromatic rings each cause predictable shifts
Predicting Spectra for Molecules Studied in the Course
This subsubtopic requires students to “predict carbon-13 or proton NMR spectra for given molecules studied in the course,” meaning students must justify:
How many NMR signals are expected
Where those signals are likely to appear
How intensity or splitting patterns arise
How functional groups modify expected values
After assigning approximate chemical shift regions, add the expected multiplicity for each proton environment and check that the integration ratio matches the proton counts.
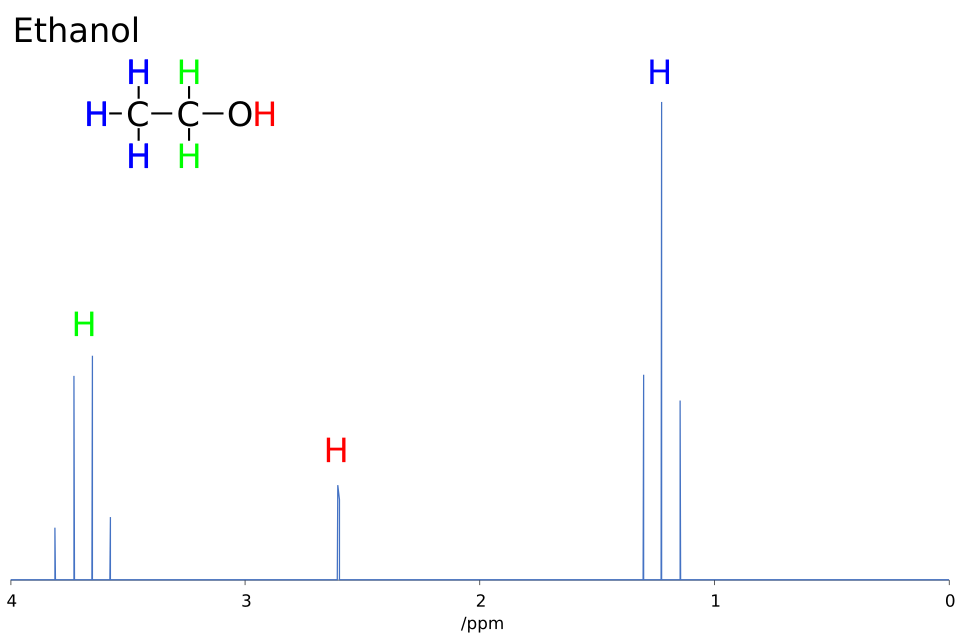
This labelled ¹H NMR spectrum of ethanol shows distinct proton environments, their chemical shifts, and the resulting splitting patterns. Expanded regions highlight how neighbouring protons generate triplet and quartet signals. Source
Bullet-Point Approach to Prediction
Students can summarise prediction reasoning as:
Identify functional groups
Locate symmetry elements
Count unique ¹³C and ¹H environments
Estimate chemical-shift ranges
Determine ¹H integration ratios
Use the n + 1 rule to predict splitting
This structured approach aligns closely with OCR expectations and ensures that predicted spectra are logical, justified, and based on fundamental NMR principles.
Practice Questions
A compound has the molecular formula C4H10O and contains no C=C bonds.
Predict the number of signals expected in its proton (¹H) NMR spectrum, stating one structural feature that justifies your answer.
(2 marks)
1 mark
Correct prediction of three proton environments / three ¹H NMR signals.
1 mark
Valid structural justification, such as:
Presence of equivalent methyl protons
Symmetry in the alkyl chain
Different environments for CH3, CH2 and O–H protons
Propanal, CH3CH2CHO, is one of the molecules studied in the course.
(a) Predict the number of signals expected in the proton (¹H) NMR spectrum of propanal.
(b) State the expected chemical-shift region for each signal.
(c) Predict the splitting pattern for each signal.
(5 marks)
(a) Number of signals (1 mark)
1 mark
Correct answer: three proton environments / three ¹H NMR signals.
(b) Chemical shift regions (2 marks)
1 mark
Aldehyde proton correctly identified at approximately 9–10 ppm.
1 mark
Alkyl protons correctly identified:
CH2 next to C=O at approximately 2–3 ppm
CH3 at approximately 0.5–1.5 ppm
(Allow credit if both alkyl regions are correctly described together.)
(c) Splitting patterns (2 marks)
1 mark
Aldehyde proton predicted as a triplet due to coupling with adjacent CH2 protons.
1 mark
Correct splitting of alkyl protons:
CH2 as a quartet (neighbouring CH3 protons)
CH3 as a triplet (neighbouring CH2 protons)
(Allow full credit if splitting patterns are correctly assigned but order is reversed.)
FAQ
Predicted chemical shifts are based on typical ranges, not exact values. Real spectra are influenced by subtle electronic effects.
Factors that can cause small differences include:
Nearby electronegative groups altering electron density
Conjugation or resonance effects
Solvent interactions
Magnetic anisotropy from aromatic rings
As a result, predicted values should be treated as approximate rather than precise.
Symmetry reduces the number of unique chemical environments in a molecule.
If atoms can be exchanged by rotation or reflection without changing the molecule, they are equivalent and give one signal.
This is particularly helpful when predicting spectra quickly, as recognising symmetry can immediately limit the number of expected peaks in both ¹H and ¹³C NMR.
Signal broadening often occurs when protons exchange rapidly or interact with neighbouring heteroatoms.
Common causes include:
O–H or N–H protons undergoing hydrogen bonding
Proton exchange with trace water or acid
Overlapping signals from similar environments
Although broadening is not always predictable in detail, students should expect exchangeable protons to appear less sharp.
Yes, particularly for simple molecules with few functional groups.
Structural isomers may share the same number of environments, similar chemical shifts, and identical splitting patterns.
This is why predicted NMR spectra are often used alongside other data, such as molecular formula or functional group tests, to confirm structures rather than identify them alone.
Coupling may be absent if protons exchange rapidly or if the coupling pathway is disrupted.
This can occur when:
Protons are attached to oxygen or nitrogen
Rapid proton exchange averages out splitting
Neighbouring protons are equivalent
Understanding these limitations helps explain why predicted splitting patterns may occasionally simplify in real spectra.

{kind=link}
{kind=link}